
In our last pieces in this series, we looked into the people involved and what must be done to get a drug to market. One begins to wonder – why does drug development necessitate all this rigour, time, and money? Understanding drug development’s history might offer context and foresight for us to see its current and future direction. As you examine the history, you’ll notice change has often been attributed to reactions to tragedies, though in many cases, a closer examination shows reforms were already underway.
We’ll also see that as international attempts to harmonise laws became successful, the regulatory histories of different countries began to overlap, especially in the second half of the 20th century. Note that the perspective below focuses on an overview primarily from a US perspective – we encourage readers to explore Dan Carpenter’s historical analysis of the FDA for a more detailed understanding and other authors for respective jurisdictions.
Contents
US: Tracing the origins of drug regulation
We didn’t know it yet, but buying drugs in the 1900s was like rolling a die. At that time, many “patent medicines” said they could fix everything from headaches to heartache. For example, Dr. D. Jayne’s Tonic Vermifuge was advertised as a cure-all for throat and respiratory diseases and a worm remedy in 1896.

An advertisement for Dr. D. Jayne’s Tonic Vermifuge
Another iconic representation of patent medicines in the 19th century was “snake oil,” touted for its supposed healing properties ranging from curing headaches to alleviating chronic pain. The lore of snake oil symbolises the archetypal patent medicine, embodying the unchecked claims and placebo remedies that were commonplace in an era before rigorous drug regulation and testing.

A photo of famous “snake liniment” flyers and bottles by Clark Stanley in the 1800s
This was the landscape before drug regulation. Imagine you’re a scientist in this narrative, eager to test and market your new drug. Maybe this lack of regulation might have felt liberating, but as time unfolds, you start seeing how the crucial need for standardisation and safety protocols becomes apparent. As time passes, evolving regulatory frameworks will increasingly shape your work.
Since the 1880s, there have been efforts to regulate the proper labelling of food and drugs and prevent contaminations in either category. Harm due to unregulated food and medicines, such as the contaminated diphtheria antitoxin incident, which was prevalent, leading to multiple children’s deaths. For the first time, the Biologics Control Act (1902) institutionalised inspections and purity testing in the US, marking a move towards formal regulation in ensuring drug safety and efficacy. Shortly after, the American Medical Association (AMA) pioneered self-regulation with a voluntary drug approval program, where companies were required to have proof of efficacy for their drugs before advertising in AMA’s journals. These set precedents for further regulatory measures like the Pure Food and Drug Act (1906) for the US Federal Drug Administration (FDA), making selling contaminated food or meat illegal. The law required truthful labelling – no one could “promise the moon and the stars” on a label anymore – at least, that was the goal.
US: Reactivity, Proactivity, and Regulatory Measures
Many regulations resulted from the public outcry that followed health disasters.
- Federal Food, Drug, and Cosmetic Act (1938): Mandated safety proof before approval, introduced following a devastating tragedy involving a poorly formulated antibiotic, leading to over a hundred deaths.
- Good Manufacturing Practices (GMP, early 1940s): Developed following an incident with a batch of sulfa drugs contaminated with the sedative phenobarbital that went awry (with hundreds of deaths and injuries).
- Nuremberg Code of 1947: Served as a guideline for clinical trials (establishing principles for ethical research like voluntary consent) in the wake of inhuman experiments by Nazis in Germany, built upon by the Declaration of Helsinki in 1964.
- FDA’s ban on GAS vaccines (1979), An arguably overreactive prohibition on using Group A Streptococcus (GAS) organisms and their derivatives in vaccines after two or three out of 21 healthy subjects developed rheumatic fever after vaccination in a study in 1969. Despite the long history of successful M protein vaccines—targeting components of bacteria like GAS—the FDA’s ban effectively stifled research in this area for nearly three decades. The decision has been considered particularly significant given the high unmet medical need related to GAS. It wasn’t until 2006 that the ban was lifted, allowing for a resumption of vital research and prospective breakthroughs.
- Faster Access to Life-saving Medications (1990s): Albeit carefully monitored, access to life-saving medications after AIDS activists criticised the FDA for blocking access to potentially life-saving drugs and slow drug approvals during the 1990s.
- The FDA Gender Guideline (1993): Required medication testing in both sexes and overturned a 1977 guideline that excluded fertile women from early clinical studies (which was a response to the Thalidomide disaster, an event described below) .
However, it’s important to note that while it is tempting to think that drug regulation emerged purely from public outcry, a sort of legislative knee-jerk reaction to high-profile disasters, that’s only part of the story. Many reforms were already simmering on the backburner before tragedy turned up the heat. Also, the policy evolution shifting from reactive to proactive regulation is notable. If legislative changes used to be more crisis-driven, today, there’s an apparent effort to anticipate risks and implement preventive measures. Voluntary guidelines have also played a role, sometimes originating from industry best practices that later become regulatory expectations.
- As early as 1933 – to exhibit shortcomings of the 1906 Pure Food and Drug Act and illustrate the need for a new law – the FDA launched the “American Chamber of Horrors,” an exhibit featuring problematic products like unsafe medical devices (e.g. harmful womb supporter and a deadly weight-loss drug) and toxic cosmetics. First Lady Eleanor Roosevelt, who had toured the exhibit, leveraged it to push for stronger consumer protections. This was a precise instance of the FDA taking the initiative to push for legislative change.
- FDA “batch certification” (Early 1940s): Mandated for certain key drugs, starting with insulin in 1941 and then penicillin in 1945. Companies were required to submit samples from each lot for FDA testing before they could be released to the market. The policy was later expanded to include all antibiotics but was discontinued in 1983, a rare example of regulations getting less stringent.
- The Belmont Report (1979): Laid out ethical standards for human subject research, emphasising respect for persons, beneficence, and justice. If you’re the scientist, the responsibility would now fall on you to ensure that every step of the drug discovery and clinical testing adheres to these ethical guidelines, a testament to the evolving integrity of the field. It profoundly impacted clinical research regulations in the US, guiding ethical considerations in medical trials. Around the same period, bioethical committees in other countries also made strides, publishing comparable guidelines and reinforcing a global commitment to ethical standards..
The Thalidomide disaster (Late 1950s – Early 1960s) is a significant chapter in drug development history, highlighting the importance of historical tension between reactivity and proactivity. Thalidomide – a drug initially approved to treat morning sickness in pregnant women, used a lot in the 1950s – led to severe congenital disabilities that were apparent in the 1960s and ignited public outcry for stronger drug laws. US Senator Kefauver, already advocating for drug reform, seized this crisis to reinvigorate a languishing bill in Congress. In July 1962, Kefauver publicised the drug’s harmful effects, breathing new life into his bill, which he felt had lost momentum.
Apart from having lots of reactions, the tragedy catalysed broad reforms in drug regulation in the US and globally. In the US, the disaster led to a mandate requiring any sponsor planning a clinical investigation of a drug to provide a detailed study outline to the FDA and demonstrate its efficacy, not just its safety. Internationally, the event spurred centralised authorisation procedures in the EU for new drug assessments. For instance, the UK passed the 1968 Medicines Act, establishing comprehensive drug classifications. Generally, the Thalidomide disaster served as a wake-up call for the industry, catalysing reforms that were not just reactive but also comprehensive and robust.
Global Harmonisation for Drug Development
The International Conference on Harmonisation (ICH) aimed to standardise the drug development process starting in the 1990s (similar to how a USB-C attempts to standardise connectors). The ICH initiative represents a collective stride towards a more streamlined and unified global drug development paradigm, ushering in a new era of collaborative efforts among drug manufacturers across borders. By bringing together regulatory bodies, from Europe, Japan, and the US, the ICH ensures that a drug tested and approved in one region can more easily gain acceptance in others, accelerating global access to vital medications.
From their website: “ICH’s mission is to achieve greater harmonisation worldwide to ensure that safe, effective, and high quality medicines are developed and registered in the most resource-efficient manner. Harmonisation is achieved through the development of ICH Guidelines via a process of scientific consensus with regulatory and industry experts working side-by-side.”
ICH has developed extensive processes for new harmonisations, clarifications, revisions, and maintenance and has ~150 Guideline documents on safety, efficacy, quality, and miscellaneous. At Step 5 of the ICH process, harmonised ICH Guidelines are implemented by ICH Regulatory Members and Observers within their respective country/region. Implementation and adherence to ICH Guidelines within Regulatory Member and Observer countries/regions are monitored via independent third-party surveys (see 2021 Project Report). ICH has a total of 21 member organisations, each with two representatives. Many regulatory authorities incorporate ICH guidelines into national law, making compliance a legal requirement.
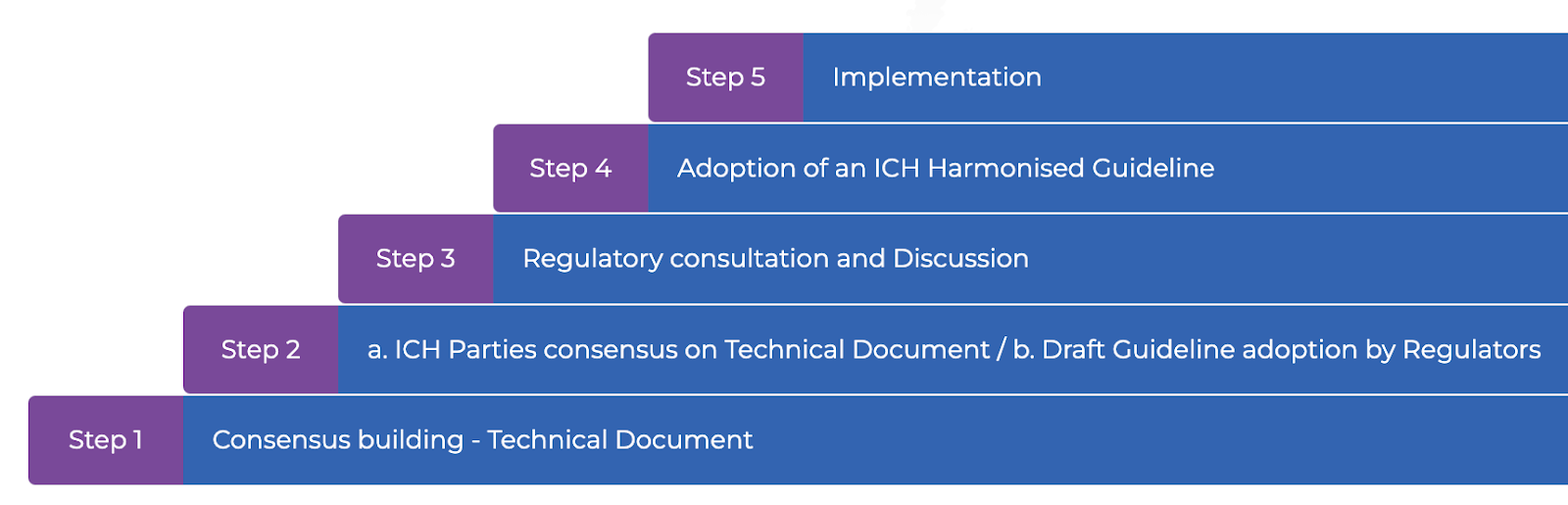
The ICH Process.
An example of the ICH process at work: As we progress into 2023, the ICH’s Good Clinical Practice (GCP) guidelines are under ongoing revision. They were initiated in 1996, last updated in 2016, and their newest revision is currently accessible as a draft. These guidelines continue to evolve, focusing on new trial types and data sources, reflecting the regulator’s and industry’s commitment to staying ahead of the curve.
Global Milestones in Regulation: A Tour from Beijing to Russia
While the history is approximately similar to the US for many Western countries, this section provides a very short peek across borders and overseas; the narrative of drug regulation also unfolds with distinct chapters written in different nations. Each country has its historical events and reforms, sculpting its regulatory landscape. This is meant to provide a concise overview and to shift away from an US centric view on drug development – we’ve linked sources for deeper dives into specific countries and don’t at all claim to have significant expertise for any of these regions. Let’s explore a few key milestones in certain countries.
Brazil
- The National Health Surveillance Agency or ANVISA (1999): Established to protect the population’s health through sanitary control.
- Law No. 13.411 (2016): Modified the pharma regulatory environment, lowering bureaucratic barriers in clinical research and improving pharmaceutical registration and post-market monitoring processes.
- Read more: Drug registration in Brazil
China
- The Pharmaceutical Company of China (1950): assigned to take charge of the nation’s wholesale trade of pharmaceuticals, at a time when hospitals/clinics were transferred from the private sector to direct government control.
- GCP Launch by the State Drug Administration or SDA (2001)
- CFDA Improvements (2015): Announced by the China Food and Medication Administration to speed up medication approval processes and give Chinese patients faster access to new drugs.
- ICH Membership (2018): China became a full member of the ICH, demonstrating its commitment to harmonising its pharmaceutical rules with global norms (and the reach of ICH).
- Read more: China’s drug regulation history
India
- Pre-Regulation
- Practicing of traditional Indian medicine systems (Pre-20th Century) Using natural ingredients like Ayurveda, Siddha, and Unani for treatment.
- Importation Era (20th Century) Most drugs were imported from Europe during their colonial period.
- The early pharmaceutical industry in India began with a few indigenous companies manufacturing basic medicines in the early 20th century, limiting local pharmaceutical development.
- Drugs and Cosmetics Act (1940): Established regulatory control over the import, manufacture, distribution, and sale of drugs and cosmetics in India, making the sale of substandard drugs a serious offence.
- In 1945, the government established the Drugs and Cosmetics Rules to classify drugs under given “schedules” and present guidelines for the storage, sale, display, and prescription of drugs.
- In 1988, the Act was reformed to integrate WHO’s GMP principles.
- In 2005, The Act’s “Schedule Y” (schedule providing clinical trial guidelines) was updated to incorporate more precise standards and instructions for conducting clinical trials in India.
- The Patents Act (1970): Allowed the development of India’s generic drug industry by recognising only process patents, not product patents.
- Modernisation and Global Expansion (2000s-Present):
- Becoming a significant player in “Contract Research and Manufacturing Services” (CRAMS)
- Formation of regulatory bodies like the Central Drugs Standard Control Organization (CDSCO) and the National Pharmaceutical Pricing Authority (NPPA) to improve drug quality, safety standards, and ensure affordability
- Changes in India’s patent laws due to the introduction of the World Trade Organization’s Trade-Related Aspects of Intellectual Property Rights (TRIPS) agreement, affecting the approach to generic drug production.
- The New Drugs and Clinical Trials Rules (2019): Reformed the regulatory framework by reducing complexity in clinical research and improving the processes for medicine registration and post-market surveillance.
Russia
- Soviet Regulation (Post-1917 Revolution – 1930): Following the 1917 Revolution, the government established a state monopoly controlling all aspects of pharmaceutical production, distribution, and quality assurance, including the creation of the Pharmacopoeia Commission in 1923 (that controlled pharmaceutical quality) and comprehensive management of pharmaceutical activities under the People’s Commissariat of Health by 1930.
- Recent regulation (2010-11): Enactment of federal laws aimed at improving healthcare and drug provision, alongside the Law “On Circulation Medicinal Products” in 2010 to ensure the quality and safety of medicines, reflecting the evolving healthcare landscape.
Zimbabwe
- The Drugs and Allied Substances Control Act (1969): An act beginning systematic control and management of medicines in the country.
- Establishment of the Medicines Control Authority of Zimbabwe or MCAZ (1997): Created to oversee and regulate medicines in the country.
- Regulations on Homeopathic Remedies (2015): An act representing an official cancellation or revocation of a previous set of regulations governing homoeopathic remedies in Zimbabwe.
- Good Distribution Practice (GDP) Guidelines Adoption (2018): Recently, the MCAZ adopted GDP Guidelines to ensure the quality and safety of medical products during all aspects of the distribution process.
What about countries that don’t have an independent drug regulatory system?
Despite these strategies, research has shown how developing countries still meet resistance in adopting global standards for pharma quality and regulatory infrastructure. Countries that don’t have their own independent regulatory system for drugs (generally smaller countries that don’t have their own explicit regulatory authorities) use certain strategies:
- WHO Prequalification: These countries regularly rely on the WHO prequalification system, which analyses and ensures the quality, safety, and efficacy of vital medications.
- The Collaborative Procedure for Accelerated Registration: spearheaded by the WHO, expedites product registration in numerous nations by leveraging the work of strict regulatory bodies.
- Reference to neighbouring frameworks: Some countries adopt or refer to the regulatory frameworks of adjacent countries or regions with more established systems.
- Reliance on international guidelines: Countries can use international guidelines from orgs like the ICH and the WHO.
Other articles in this series