
Author Note: This series was written in collaboration with James Smith and Kirsten Angeles.
Contents
Overview
In our last piece, we looked into the different stakeholders involved in drug development and the roles they play in bringing a drug from concept to market. To appreciate the meticulous nature of the journey of these stakeholders, let’s look at the actual process they participate in, starting from idea to drug.
It is commonly known that drug development happens across multiple phases and is subject to diligent protocols and documentation before market approval. While companies don’t share their R&D budgets, estimates put the cost of bringing a new drug to market from less than $1 billion to more than $2 billion. That includes the cost of failure; approximately a third of this budget is spent on candidates that never make it to market. Developing a new drug isn’t for the shallow of pocket.
In the following, we not only break down the main phases – manufacturing, discovery, preclinical testing, clinical testing, and market approval – but also want to provide a behind-the-scenes view with examples. We also go into some of the various Good Practices (GxP) guidelines – i.e., Good Manufacturing Practices (GMP), Good Laboratory Practices (GLP) and Good Clinical Practices (GCP).
| Key Aspects | Average Duration / Data | Key Regulatory Guidance | Costs and Investments | |
| Manufacturing | GMP Compliance, Quality Assurance, Record-keeping | N/A | GMP (Good Manufacturing Practices) | 15% of R&D budget; Single to double-digit million dollar runs |
| Discovery | Compound Screening, In-vitro, In-silico Testing | years | Biosafety standards | A third of overall spend on failed candidates |
| Pre-clinical Research | Pharmacokinetics, Toxicology, Pharmacodynamics, Efficacy | ~31 months on average | GLP (Good Laboratory Practices) | ~$75k for single 100 mice study, varies widely based on specifics |
| Clinical Development | Phase I to IV Trials, Trial Operations | 5.9 to 13.1 years (varies by drug type) | GCP (Good Clinical Practices), IND submission | ~$5-30M per trial on average – varies widely |
| Market Approval | NDA/BLA Submission, FDA Review | 6-10 months for first review, often with resubmissions ~400 IND applications/year; 38 new approvals/year | NDA (New Drug Application), BLA (Biologics License Application) | Approximately $2-3M as filing fee |
| Post-Market Surveillance | Post-Marketing Surveillance Reports, Efficacy Studies | Reports initially every 6 months, extending to 3+ years | Post-Marketing Surveillance Reports | N/A |
Manufacturing
Manufacturing is defined by the ICH as to “include all operations of receipt of materials, production, packaging, repackaging, labeling, relabelling, quality control, release, storage, and distribution of [drugs] and the related controls.”
The respective global standard outlining how drugs should be made and ensuring that quality is “baked into” every step of the manufacturing process – extending far beyond testing of the final product – is called GMP. In the US, it’s called Current Good Manufacturing Practices (cGMP), emphasising that manufacturers need to use up-to-date technologies and approaches to remain compliant.
In practice, this means there are requirements for every conceivable part of manufacture – look at this GMP guide to get a sense – you’ll see that even personnel hygiene has a few lines. Compliance with the requirement processes involves written protocols (e.g. in the form of SOPs, Master Formulae), employee training, internal audits, equipment testing, good quality materials being used (e.g., ensuring the traceability and quality of materials used in production), quality assurance, quality control measures, and many other things, all documented in a quality management system (QMS). GMP emphasises thorough record-keeping making tracing any activity during a manufacturing batch possible. (For more details on quality assurance, this paper provides a readable overview – it’s focused on cell therapy, but the principles are the same.)
How does GMP look in practice?
- Processes are defined and execution-checked. Every step in the manufacturing process and testing of the drug product, which consists of the actual active ingredient (drug substance) and all other adjuvants, binders, coatings, etc., must be defined by a recipe/procedure, execution against which is recorded and checked.
- This recipe will be highly detailed: for example, there might be three different steps.
- Measure 1 ml of reagent A using a specific instrument and model.
- Combine with reagent B into a specific vessel of a specific size and model.
- Record the volume of the resulting mixture using a technique defined in a standard operating procedure.
- Person A follows the procedure.
- After every small step, person A, who completed it, has to initial next to that step on a piece of paper (or electronic equivalent), confirming that they did what was stated there. If there is a result to record (e.g., as in step iii), they write that result.
- Person B observes Person A doing the step, and Person B also initials next to the step to confirm that Person A did it correctly. If Person A recorded a result, Person B confirms they recorded it correctly.
- Person A must have been trained to follow that procedure and demonstrate that they were trained to follow it. Same for Person B, the checker.
- All of this is done for every “quality-critical” step.
- This recipe will be highly detailed: for example, there might be three different steps.
- Intermediate products are tested and quality-verified. There are tests of intermediate products throughout the manufacturing process to ensure that it is within pre-defined specifications and, e.g., there has been no contamination. Often, new assays must be developed to test whether the intermediaries and final drug substance you created according to specification. It is insufficient to rely on testing the drug at the end of the process.
- Records are reviewed and approval-secured by a dedicated quality function. Before patients can use a drug,, all of the documents recording every step of the process must be reviewed by people from a department whose sole role is quality assurance.
- When all the manufacturing is completed, you have a set of documents that have been initialled by persons A and B, each one representing a part of the overall manufacturing process.
- Collectively, that set of records is referred to as the batch record. (They record the manufacture of the batch of the drug.)
- The quality function (quality assurance department) of the manufacturing facility must review the batch record.
- GMP requires that the quality function be operationally independent of the team directly doing the manufacturing to avoid conflicts of interest. The quality function typically reports to the CEO.
- In some countries, e.g., in the EU, this “batch release” can only be done by “qualified persons” (QPs) – accredited people with specific expertise in drug manufacturing.
- If a sponsor (the company who owns the drug, essentially – read more here) outsources manufacturing, this step will be done by the sponsor andthe contractor directly doing the manufacturing.
- The review will include, for example, checking that the product meets pre-defined specifications and that procedures were properly followed.
- Documents are retained for potential audits. The documents resulting from this process, and those recording the review of them that took place, will be stored securely and version controlled, and accessible to regulators during audits.
Manufacturing a drug comes with a lot of overhead. Small quantities of drug prototypes for very early experiments might be synthesised in-house by sponsors, but due to all the processes listed above, collaborations with CDMOs are often needed – and they commonly have waiting times of 6-12 months. Interestingly, only about 15% of the total R&D budget is allocated to manufacturing, with the bulk going toward quality assurance and testing. The cost definitely differs by therapeutic area, but a GMP manufacturing run often demands a single to double-digit million-dollar investment, while non-GMP batches usually are well below the million dollar mark.
Drug Discovery
There are many ways of arriving at a hypothetical candidate, some of which involve in-silico screening of thousands of compounds for efficacy depending on the therapeutic approach (e.g., High Throughput Screening (HTS), others are simple in-vitro test tubes or cell culture experiments. There is a significant reduction in the multitude of compounds as they are screened – see the following figure (source) of the failure rate at each step of drug discovery and development.
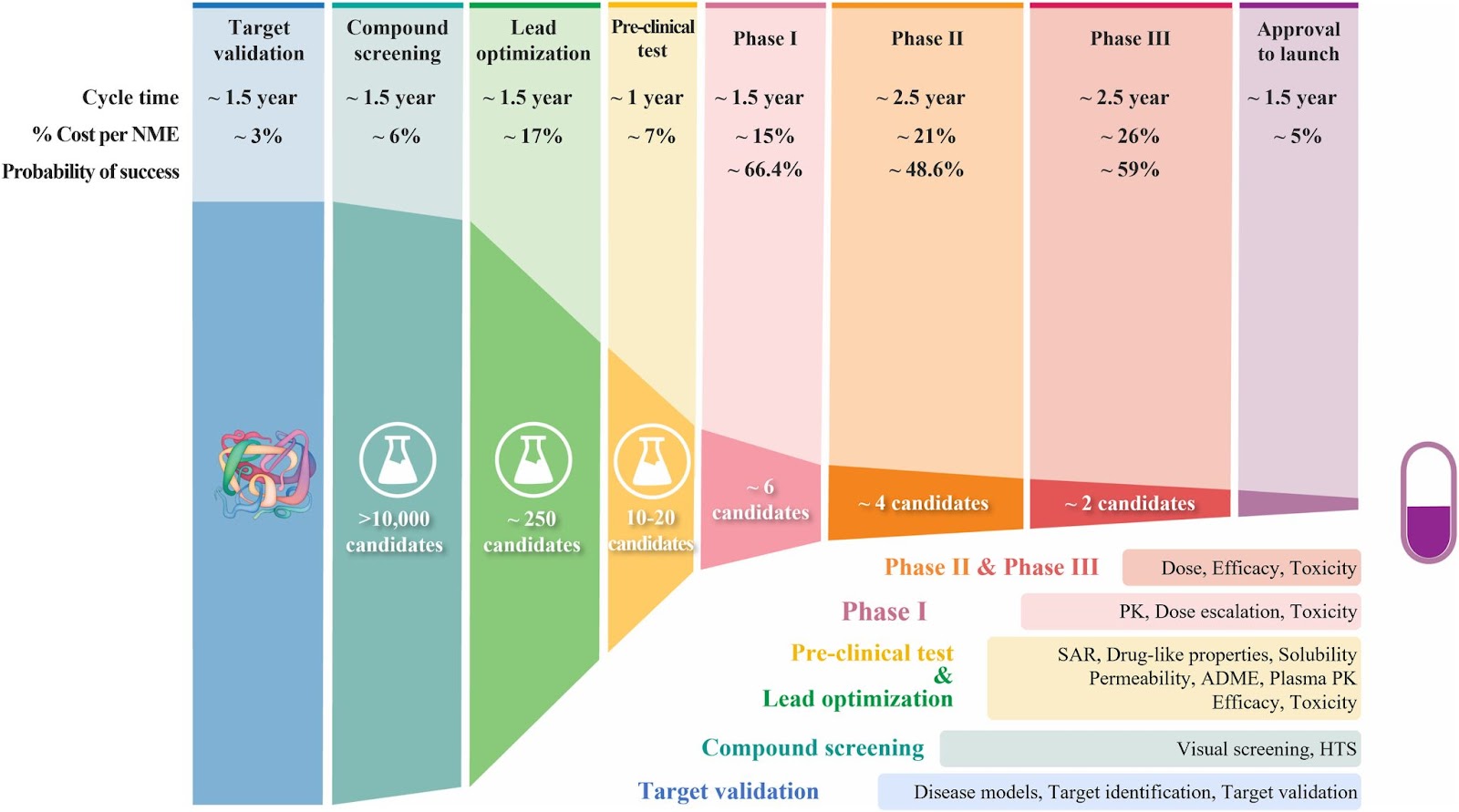
While no “Good Discovery Practices” exist and don’t require formal regulatory approval, scientists in laboratories are always subjected to biosafety standards under the purview of agencies — state agencies for lower-level labs (BSL-2 and below), and the CDC in the US for higher-level labs (BSL-3 and BSL-4). Additional regulation can apply in special circumstances, such as pathogens of specific concern (Federal Select Agent Program) or due to international collaborations.
Failed candidates consume about a third of a drug development program’s overall spend, i.e. hundreds of millions of dollars, and generally spans over the years. “Aha moments,” where scientists successfully identify compounds that target key biological mechanisms, are rarer than one would hope.
Pre-clinical research
Once a candidate is identified, it goes on to preclinical research. This stage used to require animal testing, but the FDA announced in 2022 that they now also accept non-animal organoid models.
Different parameters are studied in preclinical animal studies. While the details of how to conduct the testing of drug safety and efficacy in animals are described in GLP, with additional specifics on what to test are provided in different regulatory documents such as the Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals or the WHO guidelines on non-clinical evaluation of vaccines. In general, the following components are relevant:
- Pharmacokinetics: This area investigates the absorption, distribution, metabolism, and excretion of the drug to understand its behaviour in the biological system.
- Toxicology: Defines dose-related effects on different organ systems and developmental stages of organisms. They help to establish the safe starting dose in human trials, where the doses at the No Observed Adverse Effect Level (NOAEL) in animals get multiplied by safety factors. (A detailed elaboration on extrapolation from animal to human doses can be found here.)
- Pharmacodynamics: Determines the effects of the drug on the body, including the mechanism of action and the relationship between drug concentration and effect.
- Efficacy studies: These determine how well a drug performs its intended therapeutic function. It includes investigations into dose-response relationships to understand the optimal dosage required for the desired therapeutic effect without producing harmful side effects. This helps define the therapeutic window of a drug, a critical aspect in further clinical development.
Some of these tests can be combined; others warrant additional studies. For instance, a single toxicity study usually tests~100 animals from at least two species, and can also be used to capture additional pharmacological aspects of the drug. The total number and type of animals can rise far beyond that when additional testing in different model organisms is warranted. Depending on factors like the drug’s chemical makeup, treatment area, dose-levels, and effects being tested.
There are rarely hard requirements from regulators regarding which specific animal model to use, but there might be expectations in guidelines, and it is worth speaking to people with experience in the field (e.g., while not explicitly mentioned in guidelines, Syrian hamsters have been a standard for COVID-19 vaccine studies, sheep are a common organism for inhalation studies, and non-human primates are more and more in demand for almost any type of research, despite often being actively discouraged by regulators).
Unintuitively, the cost drivers in preclinical studies are not the number of animals but the drug itself, human labour at the study provider (number of doses, drawing blood, etc.) and analysis/pathology costs. In recent US industry quotes studies with ~100 mice with multiple injections and blood draws over 35 days, the cost per animal was around $750, totaling $75k. The price tag for manufacturing the necessary non-GMP drug can easily double this, with analysis costs ranging from 0.5 to 1.5 times the study costs. These numbers obviously can vary a lot depending on the concrete animal study plan, typically provided by the study provider once they know all the relevant variables. Preclinical testing takes about 31 months and constitutes roughly 43% of a drug development program’s total R&D spending.
Clinical development
Once a potential drug clears the preclinical studies, it moves into clinical trials in humans. Clinical development is segmented into Phase I, II, III, and IV trials, each with distinct objectives and escalating scales of operation. A single drug approval commonly involves multiple trials across different phases, testing in different patient subgroups, dose levels, or endpoints. In the EU, ~3,700 clinical trials are authorised annually, making up 46% of nearly 8,000 applications.
As a rough guide (combining different sources here, here, and here):
- Phase I: Engages up to 100 healthy participants; ~2 studies/drug development program
- Phase II: Involves ~100-500 patients and aims to establish efficacy proof of concept; ~2 studies/drug development program
- Phase III: Tested on a group of ~500-5000+ patients to generate further safety and efficacy data sufficient to convince regulators to approve the drug; ~3 studies/drug development program
- Phase IV: ~100-1500+ patients depending on the trial and drug being studied; ~3 studies/drug development program.
The total participant count of a clinical development program can range from hundreds to over 30,000 before full drug approval. Note that for certain indications, especially less common diseases, the number of patients in each phase can be much smaller than stated. The figure (source) shows the associated average costs of trial (note that substantially more expensive outliers exist)
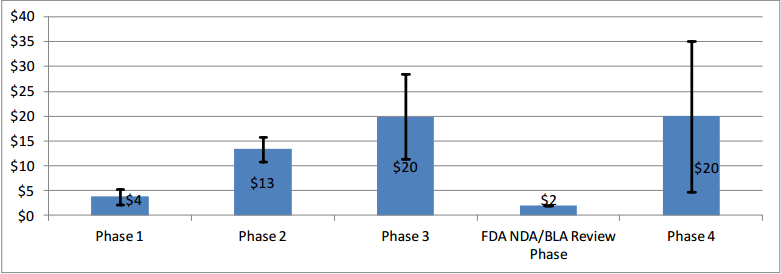
Clinical testing takes about twice as long as the discovery and preclinical stage together. Non-oncology drugs’ median duration in clinical trials ranges from approximately 5.9 to 7.2 years. For oncology drugs, that timeframe extends to 13.1 years on average. Breaking clinical development by phase, expect to spend ~1.6, 2.9, and 3.8 years in Phase I, II, and II, respectively.
The unprecedented speed of the COVID-19 vaccine development sparked hope of many for shorter timelines in the future. There often exists an impression that this is due to slow pharma companies and regulatory hurdles. Sponsor make mistakes like poor study design, ineffective site selection, poor recruitment, patient burden/safety, and trial execution and induce delays or even failures. However, it is important to note that some factors are really hard to accelerate. Sourcing certain types of participants (e.g. rare diseases) can be very challenging. While it is easy to get surrogate blood markers, endpoints we care about the most – mortality and morbidity – require longer observation periods. For instance, we could ask, “Do less people die due to cardiovascular benefits over one, three, or up to five years?” – this would be inherently impossible to expedite.
GCP is the single most important guideline for conducting clinical trials. It covers ethical considerations, informed consent, roles with their responsibilities, quality assurance and data management protocols and much more. Compliance with and training for GCP is mandatory at clinical trial sites, and principal investigators can be personally liable for non-compliance. A clear and comprehensive explanation of how all these requirements will be met must be submitted to regulators (also known as Investigational New Drug (IND) submission in the US) and local IRBs. Three of the most relevant documents are:
- The study protocol includes all primary/secondary/exploratory endpoints, statistical analysis plan, and other applicable descriptions, e.g., randomisation strategies. The clinical trial has to be performed according to protocol. Deviations from the protocol have to be recorded and can threaten the validity of the trial if mishandled. Many study protocols are published and accessible either via Clinicaltrials.gov or via the supplements of publications of the respective trial.
- The investigator’s brochure (IB) is aimed at educating physicians to conduct the clinical trial and being able to educate themselves, staff, and patients about the investigated product. This includes information on the drug’s composition and production, nonclinical study findings (usually from in vitro and in vivo testing) that demonstrate the drug’s safety profile and warrant human trials. The GCP guideline in its last chapter outlines what sections should be included in an IB. While less often publicly available, a good example is the Pfizer mRNA COVID-19 vaccine Investigator’s Brochure.
- Patient Informed Consent IRBs evaluate that human participants’ rights and well-being are safeguarded, the research’s potential benefits outweigh its possible hazards, and the investigation is carried out ethically and in accordance with recognized regulations. IRBs often provide checklists or templates what information and formatting they expect. CROs commonly know about special preferences of IRBs and can advise accordingly.
Both the regulatory authority and the IRB evaluate the document package upon receipt to assess whether the proposed trials are safe to conduct. There can be very different timelines across jurisdictions/IRBs and expert regulatory consultation can sometimes save months. Approval for a Phase 1 trial in Australia for instance can be granted in a matter of weeks, whereas the US or EU easily can take 6 months.
Similar to the GMP section, the following provides a concrete example of all activities involved in a clinical trial a sponsor needs to keep track of:
- Trial Operations
- CRO and site selection (in consultation with stakeholders – Clinical, regulatory)
- Compiling and submission of documents for CTAs
- Compiling and submission of documents for Ethics
- Study closeouts
- Project management plans for the trials, incl. trial timeline management
- High level project management of the clinical trial
- Management of vendors not associated with other SMEs
- Trial insurances
- Legal agreements related to the trial and trial vendors
- Risk log
- Action log
- Identification and Continuous collaboration with clinical trial sites and CROs
- Coordination of site visits in collaboration with relevant SMEs from Alvea
- Trial Supply Chain
- Getting the IMP to the study sites
- Procurement of Placebos / Comparators
- Management and oversight of Depots pre/during/post trial
- Ensuring adequate procurement of non-IMP supplies, either directly or in collaboration with the respective SMEs
- Data Management/processing/analysis
- Oversight to medical monitors
- IRT oversight and management
- EDC setup, monitoring, management and oversight
- Data cleaning
- Statistical analysis
- Medical Writing
- Development, revision and maintenance of
- Protocols
- IBs
- ICFs and other patient facing materials
- Lab Manuals and Pharmacy manual
- relevant additional material necessary for trial applications and execution
- Respective translations for materials when necessary
- Publication of study results
- Development, revision and maintenance of
- Participant well-being / Pharmacovigilance
- Redteaming of any safety concerns that could arise during the trial for participants
- Development, revision and maintenance of SMP / MMP
- Setup and monitoring of Safety Databases
- Safety report writing
- Management of DSMBs / IMMs
- Review and continuous monitoring of any sort of adverse events in clinical trials
- Trial testing
- Identification of capable vendors who can run testing necessary for our trial.
- Definition of the tests necessary, including physical examination, immunogenicity / safety labs, genetic testing, etc.
- Definition of materials / procedures needed for appropriate testing (e.g. tubes, swabs, measurement devices, etc.)
- Verification of the validity of the data delivered by testing vendors
- Cross-department interfaces
- Close collaboration with other departments across the whole company (e.g. manufacturing, preclinical science, operations, etc.)
- Trial Quality Assurance & Monitoring
- Oversight on development of relevant SOPs and WI in collaboration with QA team
- Verification of ongoing use of SOPs / WI across the team
- Regulatory Compliance
- Continuous mutual updating regulatory teams as well as senior leadership
- Collaboration on the definition of roadmaps for the trial team’s trajectory together with regulatory teams and senior leadership
- Informing the rest of the team about progress made between the respective parties
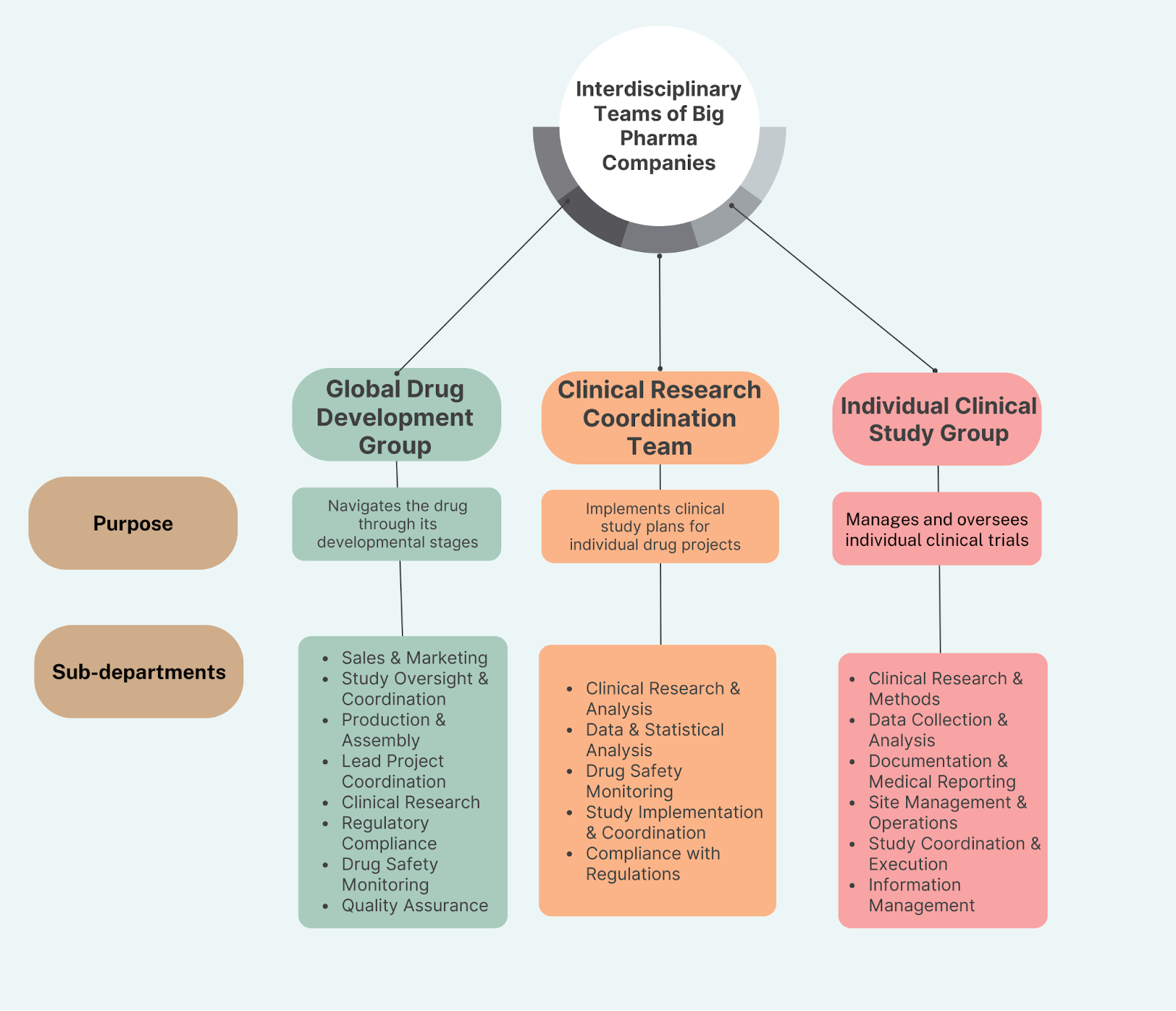
The list above illustrates that you’ll need an interdisciplinary team. For many companies, multiple of these interdisciplinary teams are stacked on top of each other:
- Global development teams think on the level of a therapeutic area (such as “Which vaccine type or blood pressure medication should we advance?”)
- Clinical program teams focus on the evaluation of drug candidates (“How do we collect all the necessary data for getting this drug candidate approved?”)
- Clinical study teams that execute individual clinical trials (“How do we get the study done efficiently and with the most valuable data?”)
Market approval
When a drug passes all these phases with good results, an application for market approval is submitted. In the US, this is submitting a New Drug Application (NDA) or Biologics License Application (BLA). Similar approval applications elsewhere are typically split into three main parts: preclinical data, detailed manufacturing methodology ensuring reproducibility, and and, of course, clinical trial information. Each section is approximately the same length, contributing to a “dossier” spanning thousands of pages. The FDA thoroughly reviews all the data and decides whether to approve the drug. This comprehensive nature is epitomised by this extensive table of contents spanning ten pages in the NDA.
Every year, the FDA processes ~400 Original Investigational New Drug applications, with an average of 38 new drugs gaining approval each year from 2010 to 2019. Of these, 3-4% are later withdrawn. Globally, the withdrawal rate hovers around 10% post-approval. Any changes to the manufacturing process, indications, or dosing require additional scrutiny and re-approval. The odds of a drug development program attaining approval is, one in seven, with rates ranging from 3.4% for oncology to a maximum of 33.4% for vaccines.
Post-Market Surveillance
The drug developer is expected by regulators to continue monitoring drug safety as it enters public use. This involves reviewing problem reports, regulating prescription drug advertisements, conducting routine inspections, etc. All data is summarised in Post-Marketing Surveillance Reports submitted by the sponsor, initially at six-month intervals, extending to three or more years as time progresses. The FDA can also require further efficacy studies while also conducting audits themselves to ensure ongoing compliance and drug safety. Both physicians and patients are encouraged to report any side effects they notice with a drug to regulatory agencies – see here for FDA’s and EMA’s contact information.
In our next piece, we will look into the origins of drug regulation, examining how historical events and key regulatory milestones have contributed to the current stringent practices in drug development. Understanding the evolution of drug regulation will provide a context to the existing rigorous processes and how they continue to shape the pharmaceutical landscape.
Other articles in this series